Regulatory Process Overview – PROCESS
Process of PMDA consultation toward CTN
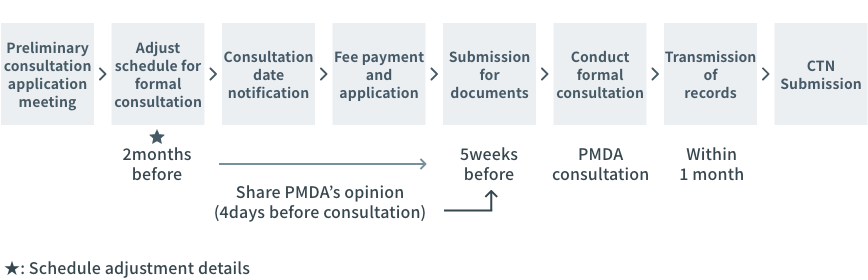
| Reception starts from 2 months before |
Date of reception start | Date of notification of consultation schedule |
|---|---|---|
| For Mar 2024 | Jan-10-2024 | Jan-16-2024 |
| For Apr 2024 | Feb-1-2024 | Feb-7-2024 |
| For May 2024 | Mar-1-2024 | Mar-7-2024 |
| For Jun 2024 | Apr-2-2024 | Apr-8-2024 |
| For July 2024 | May-8-2024 | May-14-2024 |
| For Aug 2024 | Jun-4-2024 | Jun-10-2024 |
| For Sep 2024 | Jul-2-2024 | Jul-8-2024 |
| For Oct 2024 | Aug-1-2024 | Aug-7-2024 |
| For Nov 2024 | Sep-3-2024 | Sep-9-2024 |
| For Dec 2024 | Oct-1-2024 | Oct-7-2024 |
| For Jan 2025 | Nov-1-2024 | Nov-8-2024 |
| For Feb 2025 | Dec-3-2024 | Dec-9-2024 |
| For Mar 2025 | Jan-8-2025 | Jan-15-2025 |
Ex. If the PMDA consultation is scheduled in May, it must be submitted on 1st March.
But it becomes irregular during the holiday season.
Refer to the latest notification issued by PMDA
Source: PMDA(https://www.pmda.go.jp/)
Documents required for PMDA consultation
| Content to be Included in the Documents for PMDA Consultation Meeting |
|---|
| Treatment for the relevant disease |
| Issues with existing treatments and expected benefits of the investigational drug |
| Package inserts for the US and EU and translated documents |
| Development Background |
| Complete Clinical Data Package |
| Latest Clinical Overview |
| Protocol and Investigator’s Brochure (IB) |
| List of Clinical Trials |
| List of Toxicity Study |
| Related research paper |
| Previous Consultation Records (if applicable) |
| Latest Safety Information (if applicable) |
Refer to the latest notification issued by PMDA
Source: PMDA(https://www.pmda.go.jp/)
Required documents for Clinical Trial Notification
| Required Documents |
|---|
| Document explaining the scientific rationale for the Trial |
| Protocol |
| Informed consent document |
| Sample Case Report Form |
| Latest Investigational Brochure |
| Documents detailing the latest scientific findings regarding the investigational drug use in treatments other than the investigational drug (Package Inserts, Interview Forms, Academic research papers, etc.) |
| Others |
| Documents to be submitted as needed |
|---|
| Materials related to the evaluation and management of DNA reactivity (mutagenicity) and impurities |
| Materials related to the quality of medicinal products manufactured using cultured cells, such as protein pharmaceuticals |
| Latest final report on non-clinical safety studies (toxicological and safety pharmacology studies) |
Refer to the latest notification issued by PMDA
Source: PMDA(https://www.pmda.go.jp/)
Timeline from NDA to Regulatory Approval (standard review products)
To achieve the target of 12 months (standard review products) for the total review time (median) from receipt of an application to approval, the standard review timeline indicating the timeframes (the total of the times allowed for the regulatory authorities and the applicant) for each review stage, based on past performance in regulatory review, is shown below. This timeline is applicable when there are no particular concerns in the course of review.
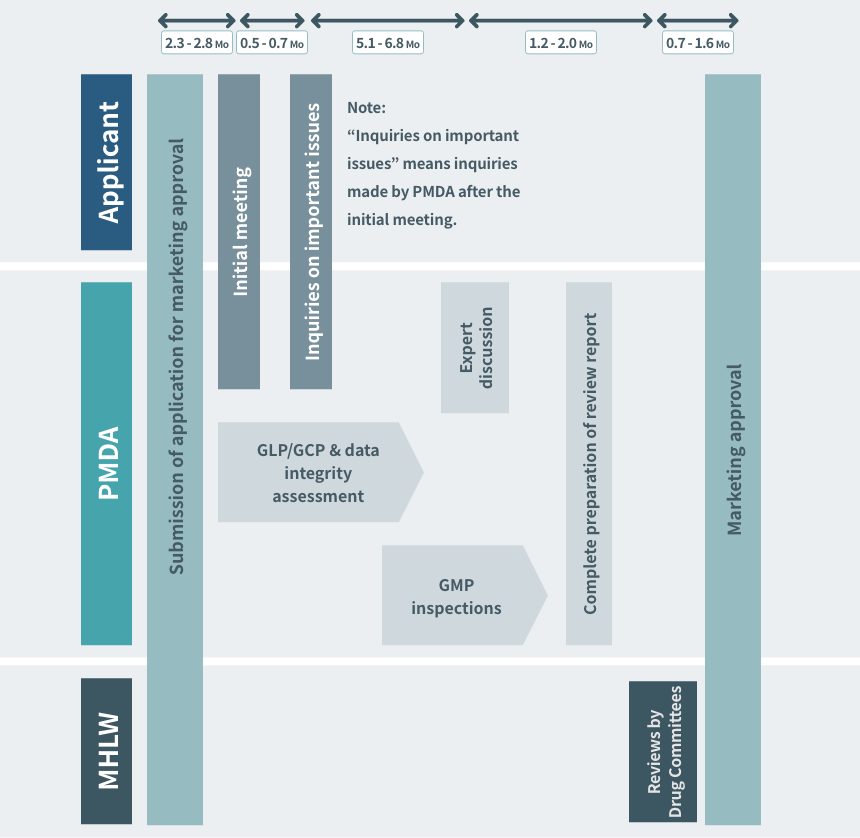
Refer to https://www.pmda.go.jp/files/000153667.pdf
Source: PMDA(https://www.pmda.go.jp/)
Timeline from NDA to Regulatory Approval (priority review products)
To achieve the target of 9 months (priority review products) for the total review time (median) from receipt of an application to approval, the standard review timeline indicating the timeframes (the total of the times allowed for the regulatory authorities and the applicant) for each review stage, based on past performance in regulatory review, is shown below. This timeline is applicable when there are no particular concerns in the course of review.
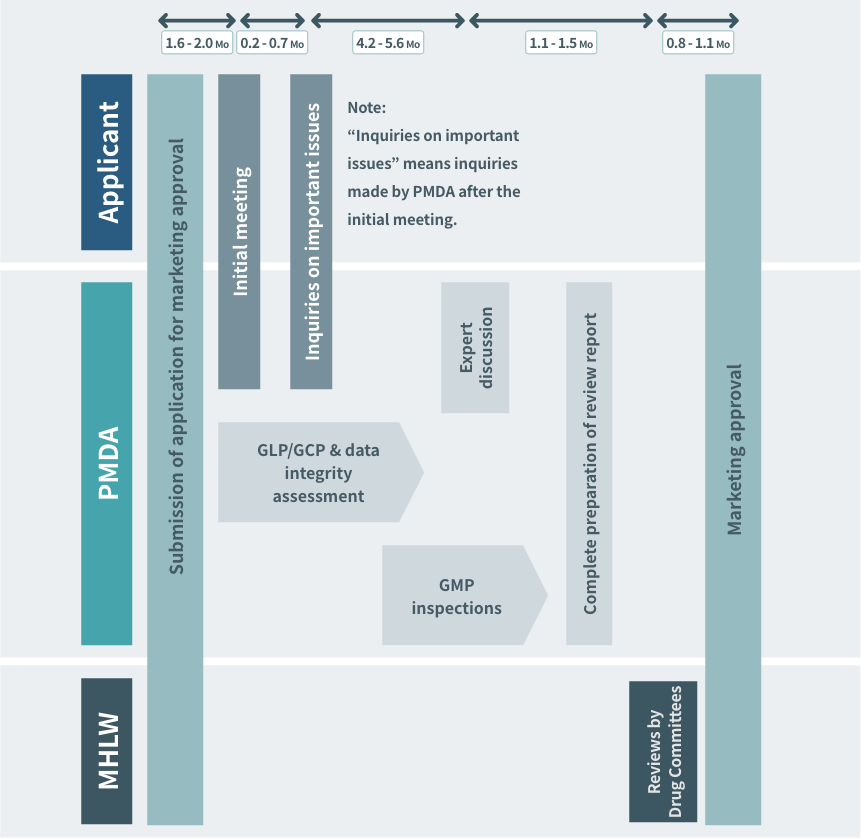
Source: PMDA(https://www.pmda.go.jp/)
Documents required for NDA
Documents to be Attached When Applying for Approval
(to be translated into Japanese)
| Specified Document | Scope of Document |
|---|---|
| ・ Origin or discovery, circumstances, and usage status in foreign countries and other related documents |
|
| ・ Manufacturing method, specifications, and test methods related documents |
|
| ・ Stability related documents |
|
| ・ Pharmacological effects related documents |
|
| ・ Absorption, distribution, metabolism, and excretion related documents |
|
| ・ Acute toxicity, subacute toxicity, chronic toxicity, mutagenicity, carcinogenicity, reproductive toxicity, and other toxicity related documents |
|
| ・ Clinical trial results related documents | Clinical Trial Results |
Refer to the latest notification issued by PMDA
Diagrammatic Representation of the ICH Common Technical Document
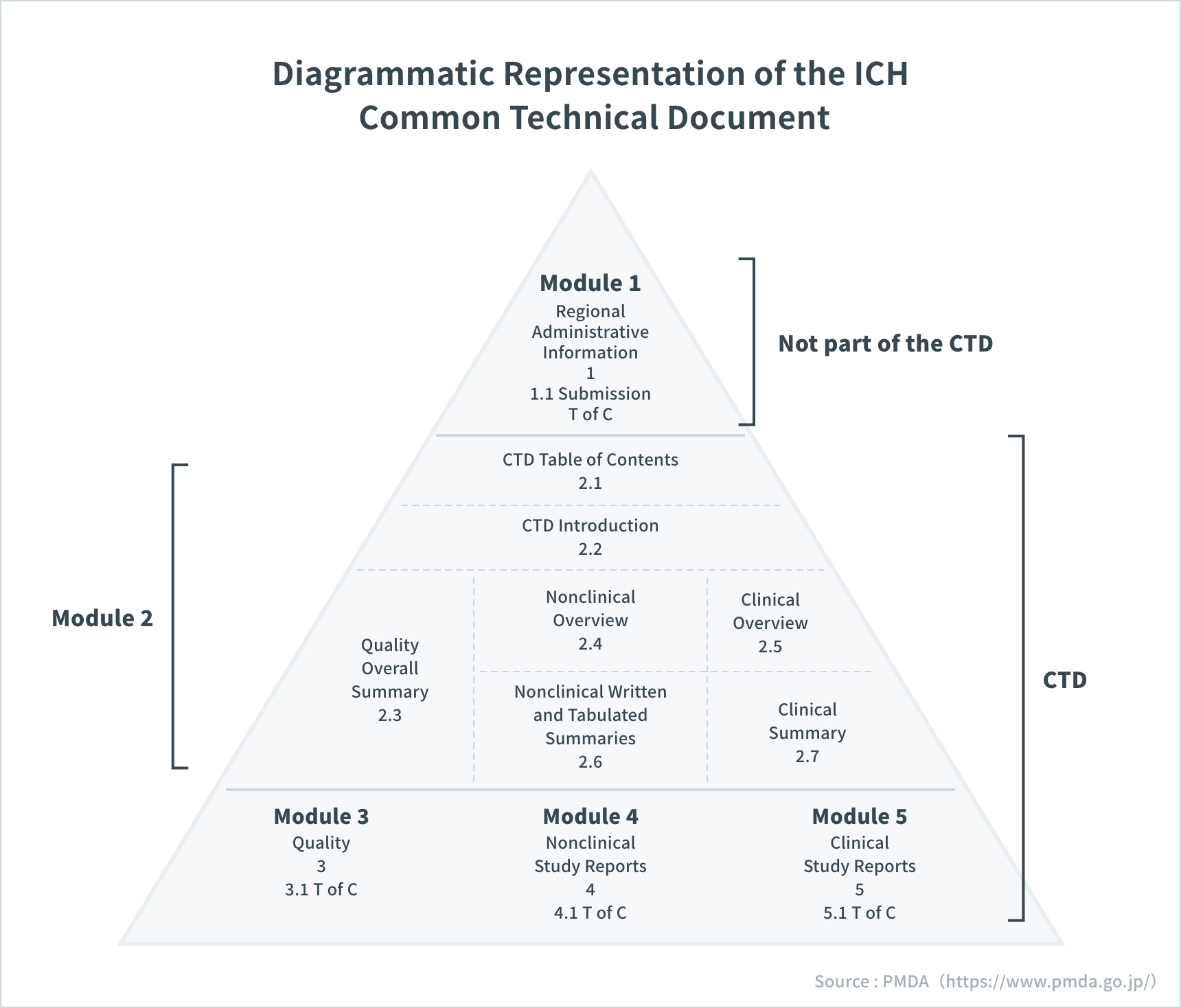
Source: PMDA(https://www.pmda.go.jp/)
Timeline (From Regulatory Approval to Drug Pricing and Marketing)
- New drug prices are listed four times a year: February, May, August, and November.
- Generic drug prices are listed in February and April.
- In some cases, drug prices are listed urgently.
- Drug prices are listed within 60 days of approval in principle, and within 90 days at the latest.

Processes of Drug Pricing
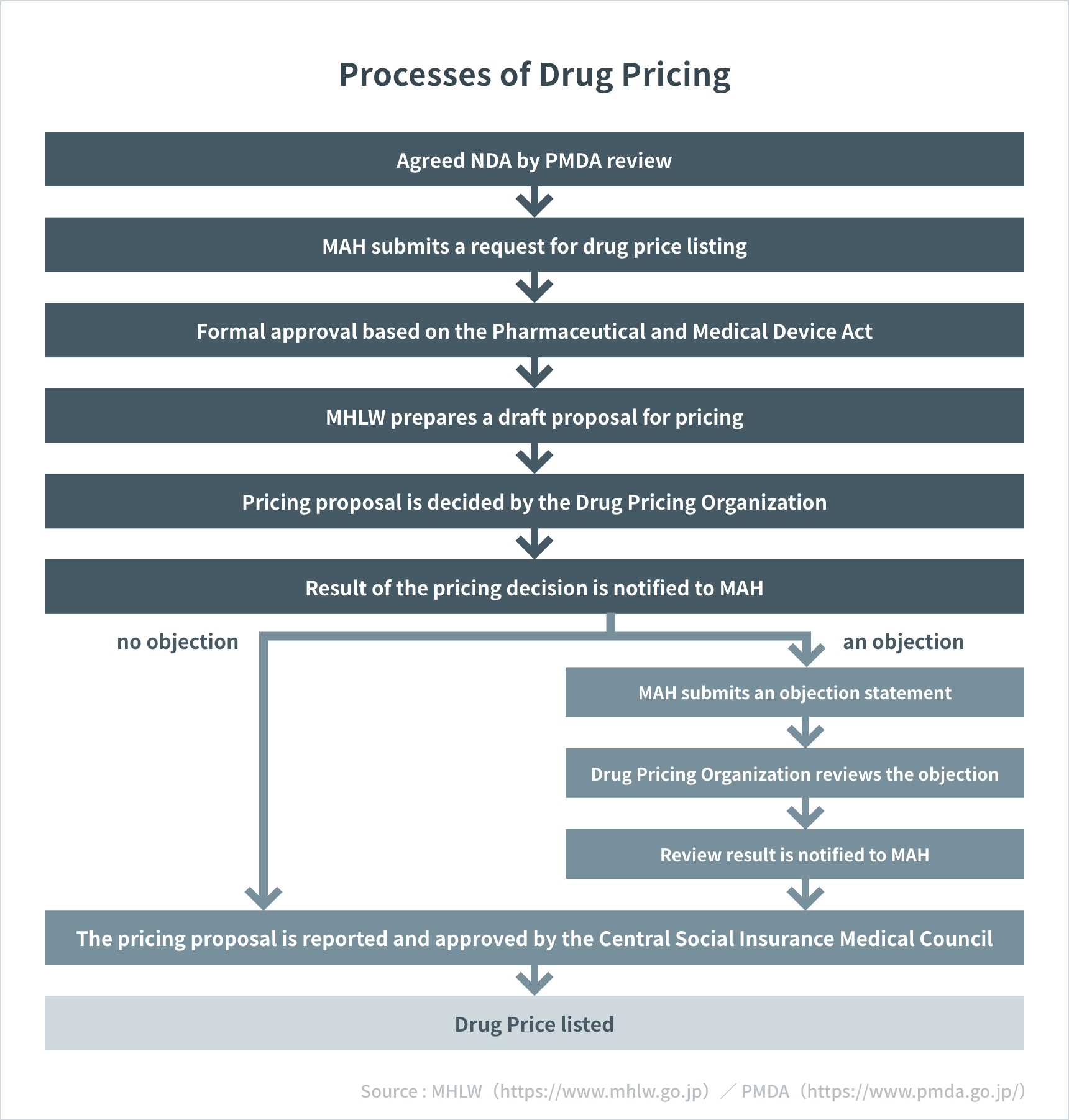
Source: MHLW(https://www.mhlw.go.jp/) / PMDA(https://www.pmda.go.jp/)
Countries which has introduced an abridged review procedure for new pharmaceutical products approved in Japan
As of Dec 2024
| Pharmaceuticals *1 | Medical devices and In vitro diagnostics *2 |
|
|---|---|---|
| European Union | ✓ | |
| Switzerland | ✓ | |
| Ukraine | ✓ | |
| Thailand | ✓ | ✓ |
| Taiwan | ✓ | ✓ |
| India | ✓ | ✓ |
| Indonesia | ✓ | |
| Singapore | ✓ | |
| Malaysia | ✓ | ✓ |
| Vietnam | ✓ | |
| Philippines | ✓ | |
| Australia | ✓ | ✓ |
| Mexico | ✓ | |
| Brazil | ✓ |
*1. The Ministry of Health, Labour and Welfare PMDA is one of the Stringent Regulatory Authorities (SRAs) defined by the WHO.
*2. The WHO Global Model Framework is the same as Japan’s medical device approval/certification system.
Source: PMDA(https://www.pmda.go.jp/)